1️⃣ Two Main Arms of Immunity
Innate Immunity = Fast + Non-specific
- First line → hours
- No memory
- Main cells → macrophages, neutrophils, NK cells
- Main proteins → complement
- Triggered by: common microbial patterns (PAMPs), damaged tissue DAMP
Adaptive Immunity = Slow + Specific
- Takes days
- Strong memory
- Main cells → T cells, B cells
- Activated by: antigen presentation by APCs
👉 Exam logic:
Innate = immediate, pattern-based
Adaptive = delayed, antigen-specific, memory-forming
2️⃣ Key Cells & Their Functions (Super High Yield)
🧱 A. Phagocytes (Innate)
Macrophages
- Long-lived, tissue-resident
- Receptors: Fcγ (IgG) + Complement (C3b)
- Functions:
- Phagocytose + kill microbes
- Present antigen to T cells → link innate ↔ adaptive
- Release cytokines (IL-1, TNF-α)

Neutrophils
- Most abundant WBC
- Short-lived (few days)
- Phagocytosis
- Kill by:
- Degranulation (enzymes, ROS)

Mast Cells & Basophils
- IgE-mediated degranulation
- Release histamine, prostaglandins, heparin
- Cause allergy + inflammation

Eosinophils
- Anti-parasite
- Attach to parasite → degranulate → toxic proteins → kill parasites

🧲 B. NK Cells = Innate + Adaptive Bridge
- Kill virus-infected + tumour cells
- Two receptors:
- CD16 (FcγRIII) → binds IgG → ADCC
- KIRs → sense absence of MHC-I → kill
- Activated by IL-2 & IFN-γ from T cells
👉 Logic:
Healthy cell = expresses MHC-I → NK inhibited
Diseased cell = low MHC-I → NK attack

3️⃣ Complement System – the Most Exam-Loved Topic



Major Functions
- Lysis (MAC C5–C9)
- Opsonisation → C3b coats bacteria → ↑ phagocytosis
- Inflammation → C3a, C5a attract neutrophils (anaphylatoxins)
Three Activation Pathways
Pathway | Trigger | Simple Memory |
Classic | Antigen + IgG/IgM | "GM makes Classic cars" |
Alternative | Direct microbial surfaces (C3b binding) | No antibody needed |
Lectin | Mannose-binding lectin on microbes | Innate antibody-free |
All pathways converge at:
👉 C3 cleavage → C5–C9 (MAC)
Clinical importance
- C3 deficiency → recurrent severe bacterial infections
- Classic pathway deficiency → autoimmune disorders
🚀 ADAPTIVE IMMUNE RESPONSE
Adaptive immunity = highly specific, has memory, long-lasting protection, mediated by B cells + T cells.
1️⃣ B CELLS — Antibody-mediated (Humoral) Immunity
What they do (high-yield):
- Develop in bone marrow
- Recognise extracellular antigens
- Present antigen on MHC-II to T helper cells
- Become plasma cells → produce antibodies
- Form memory B cells
👉 Logic:
B cell sees antigen → internalises → presents with MHC-II → T helper gives cytokines → B cell proliferates → plasma cell → antibodies.

2️⃣ ANTIBODIES (IMMUNOGLOBULINS) – ABSOLUTE MUST-KNOW

All antibodies = 2 light + 2 heavy chains
Fab = binds antigen
Fc = determines function (complement activation, opsonisation, placental transfer)
High-Yield Functions of Each Class


IgM
- First antibody in primary response
- Cannot cross placenta
- Pentamer → strongest complement activator
- Intravascular
IgG
- Most abundant (70–80%)
- Main antibody of secondary response
- Crosses placenta (IgG1, 3, 4) → fetal protection
- IgG1 & IgG3 → best complement activators + opsonisation
IgA
- Secretions (saliva, tears, GI, GU, breast milk)
- Mucosal protection by blocking microbial adhesion
- Dimer in secretions
- dimer
IgE
- Binds mast cells + basophils
- Cross-linking → allergic reactions
- Parasitic defence
IgD
- On B cell surface as a receptor
- No major serum role
👉 Memory tip:
M G A E D = MeGa Aunty Eats Doughnuts
(M = first; G = most; A = secretions; E = allergy; D = receptor)
3️⃣ VACCINATION — Why It Works
- Primary response produces memory B cells
- On re-exposure → massive, rapid IgG response
- Vaccines mimic infection without causing disease
👉 Key concept:
Vaccines keep epitopes intact but remove toxicity.
4️⃣ T CELLS — Cell-mediated Immunity

Develop: Bone marrow → thymus (selection)
Recognise antigen only when presented on MHC.
Types of T cell receptors
- αβ TCR → 95% of circulating T cells
- γδ TCR → mucosa; MHC-independent
5️⃣ THYMIC SELECTION — Extremely Exam-Loved
Positive selection
- Occurs in cortex
- TCR must recognise self-MHC → survive
Negative selection
- Occurs in medulla
- TCR binding too strongly to self-antigen → apoptosis
- Prevents autoimmunity
👉 Survivors become:
- CD4⁺ (Th cells) = MHC-II restricted
- CD8⁺ (Tc cells) = MHC-I restricted
6️⃣ CD4⁺ T CELLS (HELPER CELLS) — Commanders

Recognise antigen on MHC-II
Th1
- Produce IFN-γ, IL-2
- Activate macrophages
- Activate CD8⁺ cytotoxic T cells
- → Cell-mediated immunity
Th2
- Secrete IL-4, IL-5
- Activate B cells → antibodies
- → Humoral immunity
👉 Key polarising cytokines:
- IFN-γ + IL-12 → Th1
- IL-4, IL-5→ Th2
Other CD4 subsets (low yield but quick to remember)
- Th3 → ↑ TGF-β
- Th0 → both Th1 + Th2 cytokines
- Tr1 → ↑ IL-10 → immune suppression
7️⃣ CD8⁺ T CELLS (CYTOTOXIC T CELLS)
Recognise antigen on MHC-I
Functions:
- Kill virus-infected cells
- Kill tumour cells
- Release:
- Perforin → pores
- Granzymes → apoptosis
Types:
- Tc1 → driven by IL-12 + IFN-γ (dominant response)
- Tc2 → driven by IL-4
👉 Both kill, but differ in cytokines.

CYTOKINES
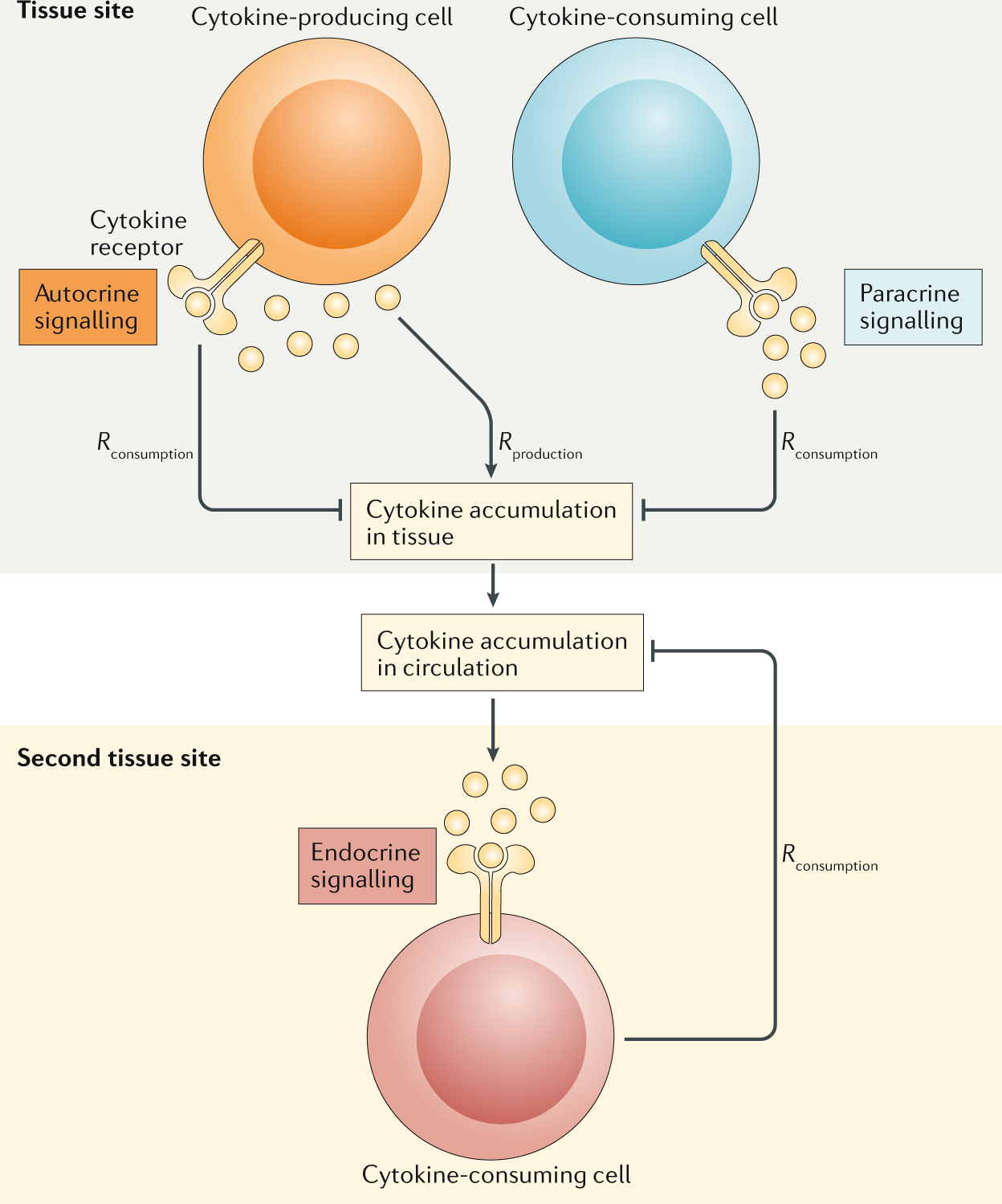
1️⃣ What are Cytokines? (Core Idea)

- Small protein messengers made by many cells: monocytes, macrophages, lymphocytes, endothelium, fibroblasts, epithelia.
- Main role: link innate and adaptive immunity.
- Act at very low concentrations via specific surface receptors.
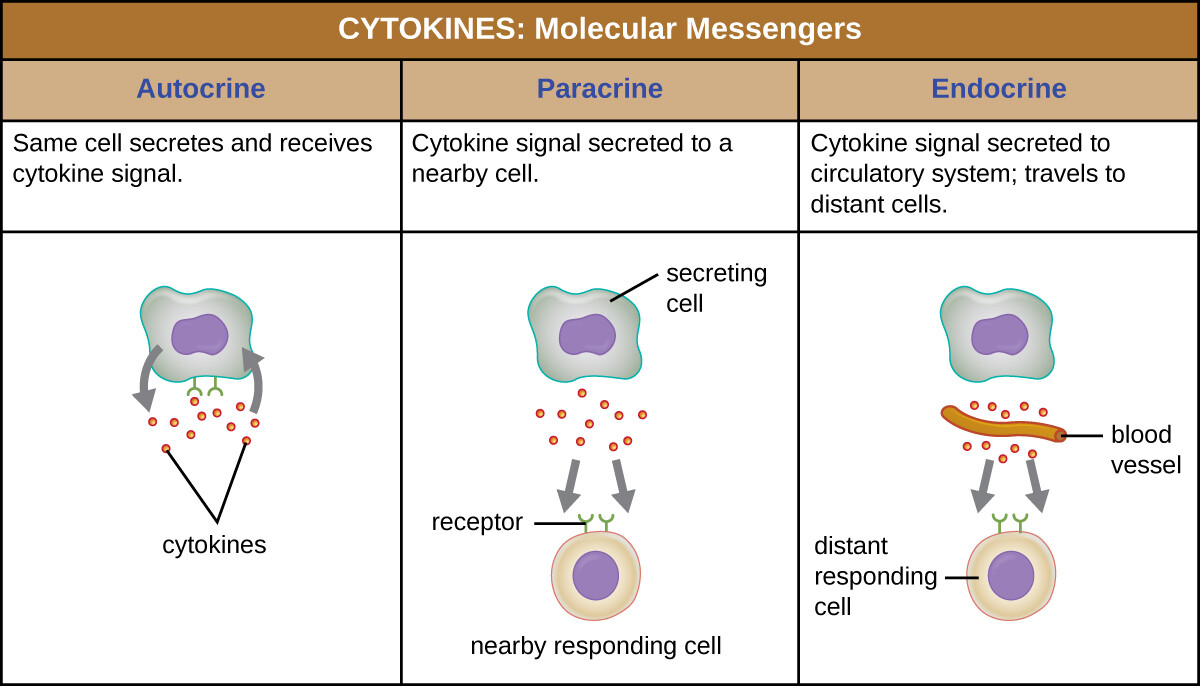
Ways they act (must know):
- Autocrine – act on same cell that secreted it.
- Paracrine – act on nearby cells.
- Endocrine – act on distant cells via blood (less common, but possible).
2️⃣ Major Cytokine Groups & Key Examples
Just know functions + sources of the big ones.
A. Interferons (IFNs)
Job: early antiviral defence until adaptive immunity kicks in.
- IFN-α – from virally infected leukocytes
- IFN-β – from virally infected fibroblasts
- IFN-γ – from Th1 cells + NK cells
- Antiviral effects
- ↑ MHC I + MHC II expression
- Activates macrophages and NK cells
- Makes target cells more susceptible to cytotoxic attack
- Pushes Th0 → Th1 (Th1 polarisation)
🔑 Functions of IFN-γ (very exam-high-yield):
B. Pro-inflammatory cytokines (the fire-starters)
Think of IL-1 and TNF-α as the first alarm signals of inflammation.
- IL-1
- Released mainly by macrophages and B cells
- Switches on immune cells like T cells, B cells, and NK cells
- Makes blood vessels express adhesion molecules, so white cells can stick and move into tissues
- TNF-α
- Released by macrophages and activated T cells
- Does almost the same job as IL-1
- Very important in the early response to infections, especially bacteria
👉 Bottom line:
When macrophages detect bacterial endotoxin (LPS) or cell stress, they release IL-1 and TNF-α to kick-start inflammation.
C.Chemokines (who-goes-where signals)
Chemokines are cytokines that act like GPS signals for immune cells.
- They pull cells toward a site by creating a chemical gradient
- Made by local tissue immune cells and endothelium
- Each immune cell has its own set of chemokine receptors, so only selected cells respond
Because of this:
- CCR5 mainly attracts Th1 cells
- CCR3 and CCR4 mainly attract Th2 cells
👉 Big picture:
Chemokines decide which type of T helper cell arrives, shaping the nature of the immune response at that site.
One-line exam lock
Chemokines don’t activate T cells — they decide which subtype shows up.
If you want, I can compress this into a single MCQ-safe sentence or a compare-with-IL-1/TNF-α block.
D. Growth factors (cell controllers)
Growth factors are signals that control blood cell formation and decide how cells mature and specialize.
- GM-CSF
- Released by macrophages, T cells, and fibroblasts
- Boosts the production of granulocytes and macrophages
- Also primes and activates these cells so they work better
- TGF-β
- Acts as an immune brake
- Slows down T- and B-cell proliferation
- Reduces activity of macrophages and natural killer (NK) cells
👉 Big picture:
GM-CSF pushes the immune response forward, while TGF-β keeps it under control.
3️⃣ MHC (HLA) – The Antigen Presentation System
T cells cannot see whole proteins.
They only respond to peptides presented on MHC molecules on APCs.
- Human MHC genes = HLA complex on chromosome 6p21.3
- Divided into:
- Class I
- Class II
- Class III
4️⃣ Class I MHC – “Show endogenous to CD8”
Types
- Classic: HLA-A, HLA-B, HLA-C
- On almost all nucleated cells
- Highly polymorphic (hundreds of alleles)
- Non-classic: HLA-E, HLA-F, HLA-G
- Limited polymorphism
- expressed only in specific tissues or cell types
- HLA-G → especially placenta (extravillous cytotrophoblast)
Function
- Present 8–9 amino acid peptides from inside the cell (endogenous antigens – e.g. viruses)
- Present to CD8⁺ cytotoxic T cells
- Allows Tc cells to kill infected cells and clear intracellular pathogens.
👉 One-liner:
Class I = all nucleated cells, endogenous peptides, CD8.
5️⃣ Class II MHC – “Show exogenous to CD4”
- Encodes HLA-DR, HLA-DQ, HLA-DP
- Expressed on professional APCs:
- Monocytes/macrophages
- B cells
- Dendritic cells
Function
- Groove is open-ended → binds longer peptides (15–24 aa)
- Presents exogenous peptides (taken up from outside the cell)
- Presents to CD4⁺ T helper cells
👉 One-liner:
Class II = APCs, exogenous peptides, CD4.
6️⃣ Class III MHC – “Support proteins nearby”
- Not peptide-presenting.
- Region contains genes for:
- Complement components: C2, C4, factor B
- Heat shock proteins: HSP70 family
- TNF-α
👉 Think: soluble defence proteins, not antigen presenters.
🧬 CYTOKINES & MHC — COMPLETE COMPARATIVE TABLES (ZERO-OMISSION)
TABLE 1️⃣ — What Are Cytokines? (Core Definition & Action Modes)
Aspect | Details |
Definition | Small protein messengers produced by immune & non-immune cells |
Main producers | Monocytes, macrophages, lymphocytes, endothelium, fibroblasts, epithelia |
Core role | Link innate immunity ↔ adaptive immunity |
Concentration | Act at very low concentrations |
Receptors | Act via specific surface receptors |
Autocrine action | Acts on same cell that secreted it |
Paracrine action | Acts on nearby cells |
Endocrine action | Acts on distant cells via blood (less common) |
TABLE 2️⃣ — Major Cytokine Groups (Overview)
Group | Main Purpose |
Interferons | Antiviral defence, immune activation |
Pro-inflammatory cytokines | Initiate inflammation |
Chemokines | Cell trafficking (who goes where) |
Growth factors | Cell production, maturation, regulation |
TABLE 3️⃣ — Interferons (IFNs)
Cytokine | Source | Key Functions |
IFN-α | Virally infected leukocytes | Antiviral defence |
IFN-β | Virally infected fibroblasts | Antiviral defence |
IFN-γ | Th1 cells, NK cells | Antiviral effects, ↑ MHC I & II, activates macrophages & NK cells, ↑ susceptibility to cytotoxic killing, Th0 → Th1 polarisation |
TABLE 4️⃣ — Pro-Inflammatory Cytokines (Early Alarm Signals)
Cytokine | Source | Key Actions |
IL-1 | Macrophages, B cells | Activates T, B, NK cells; ↑ endothelial adhesion molecules |
TNF-α | Macrophages, activated T cells | Similar to IL-1; crucial in early bacterial infection response |
Trigger for release:
Macrophage detection of bacterial endotoxin (LPS) or cell stress
TABLE 5️⃣ — Chemokines (Cell-Migration Controllers)
Feature | Details |
Core function | Create chemical gradients → attract immune cells |
Produced by | Local immune cells, endothelium |
Receptor specificity | Only cells with matching receptors respond |
CCR5 attracts | Th1 cells |
CCR3 / CCR4 attract | Th2 cells |
Big role | Decide which T-helper subtype arrives |
Exam lock | Chemokines don’t activate T cells — they select which subtype shows up |
TABLE 6️⃣ — Growth Factors
Cytokine | Source | Actions |
GM-CSF | Macrophages, T cells, fibroblasts | ↑ granulocyte & macrophage production; primes & activates them |
TGF-β | Multiple cells | Immune brake: ↓ T & B cell proliferation; ↓ macrophage & NK activity |
Big picture:
GM-CSF accelerates immunity — TGF-β restrains it
🧫 MHC (HLA) SYSTEM TABLES
TABLE 7️⃣ — MHC Overview
Feature | Details |
Human name | HLA (Human Leukocyte Antigen) |
Chromosomal location | 6p21.3 |
Function | Present peptides to T cells |
Classes | Class I, Class II, Class III |
Key principle | T cells cannot see whole proteins |
TABLE 8️⃣ — Class I MHC (“Endogenous → CD8”)
Feature | Details |
Genes (classic) | HLA-A, HLA-B, HLA-C |
Genes (non-classic) | HLA-E, HLA-F, HLA-G |
Expression | All nucleated cells |
Polymorphism | Very high (classic), low (non-classic) |
Special note | HLA-G → placenta (extravillous cytotrophoblast) |
Peptide length | 8–9 amino acids |
Antigen source | Endogenous (intracellular) |
T cell activated | CD8⁺ cytotoxic T cells |
Function | Killing infected cells |
Exam one-liner:
Class I = all nucleated cells, endogenous peptides, CD8
TABLE 9️⃣ — Class II MHC (“Exogenous → CD4”)
Feature | Details |
Genes | HLA-DR, HLA-DQ, HLA-DP |
Expression | Professional APCs only |
APCs include | Macrophages, B cells, dendritic cells |
Groove | Open-ended |
Peptide length | 15–24 amino acids |
Antigen source | Exogenous (extracellular) |
T cell activated | CD4⁺ helper T cells |
Exam one-liner:
Class II = APCs, exogenous peptides, CD4
TABLE 🔟 — Class III MHC
Feature | Details |
Antigen presentation | ❌ None |
Encoded proteins | Complement (C2, C4, factor B), TNF-α, HSP70 |
Role | Soluble immune defence proteins |
FINAL EXAM LOCK (Ultra-Short)
Cytokines signal, chemokines direct traffic, MHC shows peptides — CD8 kills, CD4 coordinates.
🚀 TRANSPLANTATION
Transplant success depends on donor–recipient compatibility, especially HLA (MHC).
1️⃣ Types of Grafts – MUST KNOW
Type | Definition | Outcome |
Autograft | Same person → self | Always accepted |
Isograft | Between identical twins | Accepted |
Allograft | Between genetically different humans | Usually rejected without immunosuppression |
Xenograft | Between species (e.g., pig → human) | Strong rejection |
👉 Exam focus:
Allografts and xenografts activate recipient T cells → rejection.
2️⃣ Allorecognition – WHY grafts get rejected
Two key reasons:
A. Passenger Dendritic Cells (Super High Yield)
- Donor dendritic cells carry donor MHC.
- They migrate out of graft → activate naïve recipient T cells.
- They are professional APCs, so the response is very strong.
B. High frequency of T cells reacting to foreign MHC
- Many recipient T cells can recognise allogeneic MHC even without peptide match.
- → Makes rejection response much stronger than against infections.
Minor Histocompatibility Antigens
- Even if MHC is identical → minor antigens can cause rejection.
- Most important: H-Y antigen (on Y chromosome) → male cells only.
3️⃣ Graft-versus-Host Disease (GVHD) – Opposite of rejection
Occurs mainly in bone marrow transplantation.
- Immune-competent donor T cells attack recipient tissues.
- Trigger against recipient MHC or minor antigens.
Prevention:
- HLA matching
- Removal of donor T cells
- Immunosuppression
👉 Exam tip:
“Rejection = host attacks graft.
GVHD = graft attacks host.”

4️⃣ Types of Rejection – The Core Exam Topic

➡️ 1. Hyperacute Rejection
Time: minutes–hours
Cause: Preformed antibodies (IgG) in recipient against donor antigens
- ABO incompatibility
- Anti-donor MHC due to previous transplant, pregnancy, transfusion
Mechanism:
Antibody binding → complement activation, thrombosis, vascular leak → graft death
👉 Immediate and irreversible.
➡️ 2. Acute Rejection
Time: days–weeks
Cause: Primary immune response
Mechanism:
- Donor “passenger leukocytes” leave graft → activate host T cells
- Host CD4⁺ (Th1) → delayed hypersensitivity
- Host CD8⁺ (Tc) → kill donor cells
👉 Reversible with immunosuppression.
➡️ 3. Chronic Rejection
Time: months–years
Mechanism:
- Progressive vascular narrowing, fibrosis
- Macrophage infiltration
- Smooth muscle proliferation
- Ischemia of graft
👉 Slowly progressive → major cause of long-term graft failure.
5️⃣ Preventing Rejection – The Only Two Ways
1. HLA Matching
- Best possible match → better graft survival
- Class II mismatches cause more severe rejection than class I
- A single class II mismatch ≈ as bad as 3–4 class I mismatches
👉 If both class I and II mismatched → very rapid rejection.
2. Immunosuppressive Drugs (Table 19.5 High Yield)

Drug | Mechanism |
Azathioprine | Inhibits nucleic acid synthesis → ↓ proliferation of all dividing cells |
Corticosteroids | General anti-inflammatory; prevent cytotoxic T cell generation |
Ciclosporin A & Tacrolimus | Block T cell activation (calcineurin inhibitors) |
Sirolimus | Blocks T cell proliferation (mTOR inhibitor) |
Anti-CTLA-4 antibodies | Promote T cell unresponsiveness to graft |
👉 Most important: Ciclosporin & Tacrolimus → T cell activation blocked.
🚀 HYPERSENSITIVITY

Hypersensitivity = harmful, exaggerated immune response → tissue damage.
There are 4 types.
Each type is defined by mechanism, immune component, and examples.
If you remember the mechanism + examples, you will ace any exam question.
1️⃣ TYPE I – Immediate (IgE-mediated)
Mechanism:
- Allergen enters via inhalation/ingestion
- Binds IgE already attached to mast cells/basophils
- Cross-linking → mast cell degranulation
- Releases histamine → vasodilation, bronchoconstriction → allergy symptoms
- INCREASE TRIPTASE
Timing: seconds to minutes
Examples:
- Anaphylaxis (peanuts, bee venom, penicillin)
- Asthma
- Hay fever
- Urticaria (weal and flare reaction)
👉 Key idea: IgE + mast cells → histamine → immediate reaction.
2️⃣ TYPE II – Antibody-mediated (IgG/IgM)
Mechanism:
- IgG or IgM bind to antigens on host cells
- Antibodies recruit:
- Complement
- Neutrophils
- Platelets
- → cell destruction + inflammation
Timing: minutes to hours
Examples:
- Transfusion reactions (wrong blood group)
- Haemolytic disease of the newborn (Rh incompatibility)
- Certain drug-induced haemolysis
- hashimotos thyroiditis
- rheumatic fever
👉 Key idea: Antibody targets cells → destruction.
3️⃣ TYPE III – Immune Complex–mediated
Mechanism:
- Failure to clear antigen–antibody complexes
- Complexes deposit in tissues (vessels, joints, kidneys)
- Activate complement → recruit neutrophils
- Neutrophils release enzymes → tissue damage
Examples:
- Autoimmune diseases:
- SLE
- Rheumatoid arthritis
- Chronic infections:
- Leprosy
- Viral hepatitis
- Hypersensitivity pneumonitis:
- Farmer’s lung
- Pigeon fancier’s lung
- Serum sickness
👉 Key idea: Immune complexes stick to tissues → complement → inflammation.
4️⃣ TYPE IV – Delayed (T-cell mediated)
Mechanism:
- Sensitised T cells (CD4 or CD8) respond to antigen at site
- Reaction develops over 24–72 hours
- No antibodies involved
- 3 forms:
A. Contact dermatitis
- Nickel, chromate
- Eczema-like rash at site
B. Tuberculin-type reaction
- PPD test (Mantoux)
- Local swelling from memory T cells
C. Granulomatous type
- Persistent antigen → chronic T-cell activation
- Cytokines (TNF-α) → granuloma formation
- Seen in:
- TB
- Leprosy
- Crohn’s disease
👉 Key idea: T-cells cause inflammation → delayed onset.
🧠 HYPERSENSITIVITY — COMPLETE MASTER TABLE (ZERO OMISSION)
Feature | TYPE I – Immediate | TYPE II – Antibody-mediated | TYPE III – Immune Complex | TYPE IV – Delayed (Cell-mediated) |
Alternate name | Immediate / Atopic / IgE-mediated | Cytotoxic hypersensitivity | Immune-complex hypersensitivity | Delayed-type hypersensitivity (DTH) |
Primary immune component | IgE antibodies | IgG / IgM antibodies | IgG / IgM immune complexes | T lymphocytes (CD4⁺ / CD8⁺) |
Antibody involved? | ✅ Yes (IgE) | ✅ Yes (IgG, IgM) | ✅ Yes (IgG, IgM) | ❌ No antibodies |
Main effector cells | Mast cells, basophils | Neutrophils, macrophages, NK cells, complement | Neutrophils, complement | T cells, macrophages |
Target | Free allergen | Antigens on host cells or ECM | Soluble antigen–antibody complexes | Antigen-presenting cells / infected cells |
Key initiating event | Allergen cross-links IgE on mast cells | Antibody binds cell-surface antigen | Failure to clear immune complexes | Sensitised T cells re-exposed to antigen |
Core mechanism | Mast-cell degranulation | Antibody-mediated cell injury | Immune-complex deposition + inflammation | Cytokine-mediated inflammation or cytotoxicity |
Major mediators | Histamine, leukotrienes, prostaglandins | Complement (C3a, C5a), ROS, enzymes | Complement, neutrophil enzymes | IFN-γ, TNF-α, IL-2 |
Complement activation | ❌ No | ✅ Yes | ✅ Yes | ❌ No |
Pathological result | Vasodilation, bronchoconstriction, edema | Cell lysis, opsonization, inflammation | Vasculitis, arthritis, nephritis | Tissue inflammation, granuloma formation |
Timing of reaction | Seconds–minutes | Minutes–hours | Hours–days | 24–72 hours |
Local vs systemic | Both | Both | Usually systemic (can be local) | Usually local |
Reversibility | Usually reversible | Often irreversible cell damage | Chronic, progressive | Chronic if antigen persists |
📌 CLASSIC EXAMPLES (HIGH-YIELD)
Type | Diseases / Examples |
TYPE I | Anaphylaxis (peanuts, bee venom, penicillin), Asthma, Allergic rhinitis (hay fever), Urticaria (weal & flare), Food allergy |
TYPE II | Blood transfusion reaction, Hemolytic disease of the newborn (Rh), Autoimmune hemolytic anemia, Hashimoto thyroiditis, Rheumatic fever, Drug-induced hemolysis |
TYPE III | SLE, Rheumatoid arthritis, Serum sickness, Post-streptococcal glomerulonephritis, Polyarteritis nodosa, Farmer’s lung, Pigeon fancier’s lung, Chronic hepatitis |
TYPE IV | Contact dermatitis (nickel, chromate), Tuberculin (Mantoux) test, TB granuloma, Leprosy, Crohn’s disease, Type 1 diabetes mellitus, Graft rejection |
🧠 EXAM LOCK — ONE-LINE MEMORY KEYS
- Type I: IgE + mast cell + histamine → immediate allergy
- Type II: Antibody attacks host cell
- Type III: Immune complexes deposit in tissues
- Type IV: T-cells only → delayed reaction
⚠️ COMMON EXAM TRAPS (DON’T MISS)
- Mantoux test = Type IV (NOT antibody-mediated)
- SLE = Type III (immune complexes, NOT Type II)
- Anaphylaxis = Type I (IgE)
- Granuloma formation = Type IV
- Complement activation = Types II & III only
fetus as an allograft
1️⃣ Core Idea: Why Doesn’t the Mother Reject the Fetus?
- Fetus is half paternal → immunologically like a semi-allograft.
- But normally not rejected → because:
- The placenta is the real interface, not the fetus directly.
- The placenta is designed immunologically to avoid classic graft rejection and to actively promote tolerance.
2️⃣ Two Maternal–Fetal Interfaces (Only the Essence)
Interface 1 – Tissue–Tissue (Decidua ↔ Extravillous Cytotrophoblast)
- Extravillous cytotrophoblast:
- Invades decidua + spiral arteries → remodels them → ↑ blood flow.
- Decidua immune cells ≈ 40% of cells:
- Majority = special NK cells (decidual NK, CD56⁺ bright, no CD16⁻)
- Some T cells, macrophages, dendritic cells
- Virtually no B cells
👉 These NK cells are not killers here – they mainly secrete cytokines, chemokines, angiogenic factors that help trophoblast invasion and placentation.
Interface 2 – Tissue–Blood (Syncytiotrophoblast ↔ Maternal Blood)
- Syncytiotrophoblast lines villi and is bathed in maternal blood.
- In contact with all maternal blood cells.
- Sheds:
- Microparticles, DNA, mRNA into maternal blood
- Occasional fetal RBCs and leukocytes → can reach maternal circulation (basis for immunisation, Rh disease).

3️⃣ Crucial MHC Pattern on Trophoblast (EXAM GOLD)
Extravillous Cytotrophoblast (Interface 1)
- Class I positive but with a very special pattern:
- Does NOT express: HLA-A, HLA-B (highly polymorphic, main graft rejection drivers)
- DOES express:
- Classic: HLA-C (polymorphic)
- Non-classic: HLA-E, HLA-G (low polymorphism)
Syncytiotrophoblast (Interface 2)
- No class I MHC at all (almost unique, like RBCs).
- No class II either.
- So it doesn’t present antigen to maternal T cells → unlikely to trigger classic T cell–mediated rejection.
Class II
- Neither trophoblast type expresses class II → again reduces T cell activation.
👉 Key exam line:
Placenta avoids classic T-cell rejection by:
- Not expressing HLA-A/B or class II
- Using HLA-C, -E, -G mainly to signal to NK cells, not T cells.

4️⃣ HLA-G, HLA-E, HLA-C – What You Actually Have to Know

HLA-G (Super High Yield)
- On extravillous cytotrophoblast.
- Low polymorphism → paternal HLA-G ≈ maternal HLA-G → less likely to activate alloreactive T cells.
- Has membrane forms (e.g. HLA-G1) and soluble forms (e.g. HLA-G5).
Functions:
- Can bind peptides, but probably mainly for molecule stability, not broad antigen presentation.
- Acts on CD4⁺ and CD8⁺ T cells:
- Induces apoptosis of CD8⁺ T cells (via Fas–FasL)
- Suppresses CD4⁺ T-cell and Tc proliferation
- Binds NK and myeloid receptors:
- KIR2DL4, ILT-2, ILT-4 on NK cells, monocytes, macrophages, dendritic cells
- Result = not attack, but support implantation:
- Decidual NK cells produce:
- Cytokines: IFN-γ, IL-10, TGF-β1
- Chemokines: IL-8, IP-10
- Angiogenic factors: VEGF, PlGF(placental growthfactor)
→ Enhance trophoblast invasion + vascular remodelling
HLA-E
- Also on extravillous trophoblast and many other cells.
- Low polymorphism.
- Binds peptides from leader sequences of other class I molecules (HLA-A/B/C/G).
- Interacts with NK receptors → contributes to NK modulation, not classic T-cell-driven rejection.
HLA-C
- Polymorphic, in theory could provoke T-cell rejection.
- But here the main interaction is again with NK cells, via KIRs.
- NK–HLA-C cross-talk controls cytokine and angiogenic factor production.
👉 Key clinical point:
Specific HLA-C / KIR combinations can promote or inhibit invasion:
- HLA-C1 + activating KIR-B on NK → good invasion → normal placentation
- HLA-C2 + inhibitory KIR-A → poor NK activation → shallow invasion → pre-eclampsia/recurrent miscarriage association
5️⃣ Maternal Antibody Responses – Why They Usually Don’t Harm the Fetus
- Fetal leukocytes (with HLA-A, -B, -C) can enter maternal blood.
- Mother can form antibodies to paternal HLA:
- About 15% of first pregnancies
- About 60% of later pregnancies with same father
- These antibodies usually don’t harm the fetus because:
Placental “Filter” Mechanism:
- Only IgG crosses the placenta (via Fc receptors on syncytiotrophoblast).
- Potentially harmful anti-paternal HLA antibodies:
- cross syncytiotrophoblast
- but then bind to HLA on villus macrophages/endothelium
- form immune complexes, which are:
- cleared by villous macrophages
- protected from complement by regulatory proteins (e.g. decay-accelerating factor)
→ So they don’t reach fetal circulation in destructive form.
Exception: Haemolytic Disease of the Newborn (RhD)
- Here mother makes IgG against fetal RBC antigen (RhD).
- IgG crosses placenta → binds fetal RBCs → haemolysis.
- Typically:
- First RhD⁺ baby of RhD⁻ mother usually okay
- Sensitisation at delivery → memory B cells
- Subsequent RhD⁺ babies → hemolysis, anaemia, liver/spleen dysfunction, can be fatal.
- Prevention: Anti-D prophylaxis postpartum:
- Inject anti-RhD IgG → coat fetal RBCs in mum’s blood → clear them before she becomes sensitised.

6️⃣ Th1/Th2 Shift in Pregnancy – KEY CONCEPT
- Normal pregnancy = bias towards Th2 (antibody) and away from Th1 (cell-mediated) responses.
Mechanism:
- Placenta produces:
- IL-4, IL-10 → Th2-promoting cytokines
- Progesterone → inhibits Th1, including IFN-γ production.
Effect:
- Cell-mediated (Th1) responses suppressed
- Humoral (Th2) responses preserved → infection defence via antibodies maintained.
Clinical evidence:
- Rheumatoid arthritis (Th1-mediated) → often improves in pregnancy.
- Diseases with intracellular pathogens (e.g. herpes, malaria) → worsen (need Th1).
- SLE (Th2-driven autoantibody disease) → often worsens in pregnancy.

7️⃣ When Things Go Wrong – Immune Mechanisms in Pregnancy Disorders
A. Abnormal HLA-G / HLA-C Patterns
- ↓ HLA-G on extravillous trophoblast → linked to recurrent miscarriage, pre-eclampsia.
- HLA-C2 + inhibitory KIR-A combinations → poor trophoblast invasion → more common in pre-eclampsia, recurrent miscarriage.
B. Th1/Inflammatory Shift in Pathology
- In pre-eclampsia/recurrent miscarriage:
- Higher IFN-γ and inflammatory markers (e.g. CRP)
- Stronger systemic inflammatory response
- Leads to endothelial dysfunction: hypertension, proteinuria, oedema, DIC.
C. Antiphospholipid Antibodies
- Lupus anticoagulant and anticardiolipin antibodies → ↑ miscarriage risk.
- Interfere with:
- Coagulation (prothrombin → thrombin)
- Trophoblast maturation + placentation
- Treatment: Low-dose aspirin ± heparin can improve outcomes.
- They can cross placenta → transient neonatal autoimmune issues.

FETUS AS AN ALLOGRAFT — COMPLETE INTEGRATED TABLE (ZERO OMISSION)
Domain | Component / Feature | Exact Details (Fully Integrated) | Exam / Clinical Lock |
Core Concept | Nature of fetus | Fetus is semi-allogeneic (½ paternal antigens) → immunologically resembles a semi-allograft | Despite this, normal pregnancy is not rejected |
Why rejection does not occur | Immune interaction is placenta-mediated, not direct fetal tissue exposure | Placenta is an active immunological organ | |
Overall strategy | Avoid classical graft rejection + actively induce tolerance | Not passive immune ignorance | |
--- | --- | --- | --- |
Maternal–Fetal Interfaces | Interface 1 | Tissue–Tissue interface: Decidua ↔ Extravillous cytotrophoblast (EVT) | Dominated by NK–trophoblast cross-talk |
Interface 2 | Tissue–Blood interface: Syncytiotrophoblast ↔ maternal blood | Avoids antigen presentation entirely | |
--- | --- | --- | --- |
Interface 1 (Decidua ↔ EVT) | Trophoblast type | Extravillous cytotrophoblast | Invades decidua + spiral arteries |
Function | Spiral artery remodeling → ↓ resistance → ↑ uteroplacental blood flow | Failure → pre-eclampsia | |
Decidual immune cells | ~40% immune cells | Unique immune microenvironment | |
Dominant immune cell | Decidual NK cells 70%(CD56⁺⁺ bright, CD16 Negative) | NOT cytotoxic here | |
Other immune cells | T cells10%, macrophages 20%, dendritic cells | Virtually no B cells | |
NK cell behavior | Secrete cytokines, chemokines, angiogenic factors | Promote placentation | |
NK secretions | IFN-γ, IL-10, TGF-β1; IL-8, IP-10; VEGF, PlGF | Aid invasion + vascular remodeling | |
--- | --- | --- | --- |
Interface 2 (Syncytiotrophoblast ↔ Blood) | Cell type | Syncytiotrophoblast | Multinucleated, lines villi |
Exposure | Direct contact with maternal blood cells | Highest immunologic risk zone | |
Shedding | Microparticles, fetal DNA, mRNA | Basis of NIPT | |
Fetal cells entering mother | Occasional fetal RBCs + leukocytes | Basis for Rh immunisation | |
Immune strategy | No antigen presentation | Immune invisibility | |
--- | --- | --- | --- |
MHC Expression – Core Exam Area | EVT – Class I | Present, but highly selective | Immune modulation, not rejection |
EVT – Absent MHC | HLA-A, HLA-B absent | These are main graft-rejection drivers | |
EVT – Present MHC | HLA-C (classic) + HLA-E, HLA-G (non-classic) | NK-directed signaling | |
Syncytiotrophoblast | No class I, no class II MHC | Almost unique (RBC-like) | |
Class II (both) | Absent on all trophoblast | No CD4⁺ T-cell activation | |
Key exam line | Placenta avoids rejection by lacking HLA-A/B + class II and using HLA-C/E/G | Repeat verbatim | |
--- | --- | --- | --- |
HLA-G (Highest Yield) | Location | Extravillous cytotrophoblast | Implantation zone |
Polymorphism | Low polymorphism | Paternal ≈ maternal | |
Forms | Membrane (HLA-G1), soluble (HLA-G5) | Soluble immunosuppression | |
T-cell effects | Induces CD8⁺ apoptosis (Fas–FasL); suppresses CD4⁺/CD8⁺ proliferation | Direct tolerance | |
NK / myeloid receptors | KIR2DL4, ILT-2, ILT-4 | Inhibitory + modulatory | |
Net effect | NK cells support implantation, not kill | Key conceptual pivot | |
--- | --- | --- | --- |
HLA-E | Expression | EVT and many somatic cells | Low polymorphism |
Peptide source | Leader peptides from HLA-A/B/C/G | Stabilisation | |
Function | NK modulation | Secondary tolerance signal | |
--- | --- | --- | --- |
HLA-C | Nature | Polymorphic | Potential T-cell risk |
Main interaction | NK cells via KIRs, not T cells | Functional re-routing | |
Role | Regulates cytokine + angiogenic output | Determines invasion depth | |
Good combo | HLA-C1 + activating KIR-B | Normal placentation | |
Bad combo | HLA-C2 + inhibitory KIR-A | Pre-eclampsia, miscarriage | |
--- | --- | --- | --- |
Maternal Antibodies | Exposure | Fetal leukocytes enter maternal blood | Alloimmunisation possible |
Anti-paternal HLA Ab | ~15% first pregnancy, ~60% later pregnancies | Usually harmless | |
Why harmless | Placental filtering + immune complex clearance | Not fetal-destructive | |
Ig class crossing placenta | Only IgG (via Fc receptors) | IgM cannot cross | |
Placental protection | Binding to villous macrophages/endothelium + complement regulators (DAF) | Immune neutralisation | |
--- | --- | --- | --- |
Exception | Rh disease | Maternal anti-RhD IgG | Targets fetal RBCs |
Timing | First RhD⁺ usually safe → sensitisation at delivery | Memory response | |
Subsequent fetus | Hemolysis, anemia, hydrops, death | High-yield | |
Prevention | Anti-D IgG postpartum | Clears fetal RBCs | |
--- | --- | --- | --- |
Th1 / Th2 Shift | Pregnancy bias | Th2 dominant, Th1 suppressed | Core physiology |
Placental cytokines | IL-4, IL-10 | Th2 skew | |
Hormonal effect | Progesterone inhibits IFN-γ | Suppresses Th1 | |
Functional result | ↓ cell-mediated immunity, preserved antibodies | Infection trade-off | |
Disease behavior | RA improves; SLE worsens; intracellular infections worsen | Exam classic | |
--- | --- | --- | --- |
Pathology – Immune Failure | HLA-G deficiency | ↓ EVT tolerance | Miscarriage, pre-eclampsia |
KIR–HLA mismatch | HLA-C2 + KIR-A | Shallow invasion | |
Th1 dominance | ↑ IFN-γ, CRP | Systemic inflammation | |
Clinical effects | Endothelial dysfunction → HTN, proteinuria, DIC | Pre-eclampsia | |
Antiphospholipid Abs | LA, anticardiolipin | Placental thrombosis | |
Treatment | Low-dose aspirin ± heparin | Improves outcomes | |
Neonatal effect | Transient autoimmune features | IgG mediated |
One-Line Exam Reflex (Lock It):
The fetus survives as a semi-allograft because the placenta avoids classical T-cell recognition (no HLA-A/B or class II), actively modulates NK cells via HLA-C/E/G, biases immunity toward Th2, and filters maternal antibodies.